

US and Chinese legislation
US legislation

What is the framework of US Legislation?


Figure 1: Framework of FDA Legislation. Adapted from FDA Legislation. Copyright 2020, Kvalito Consulting Group.
How to classify a device under US regulation?

How to classify a device under US regulation?
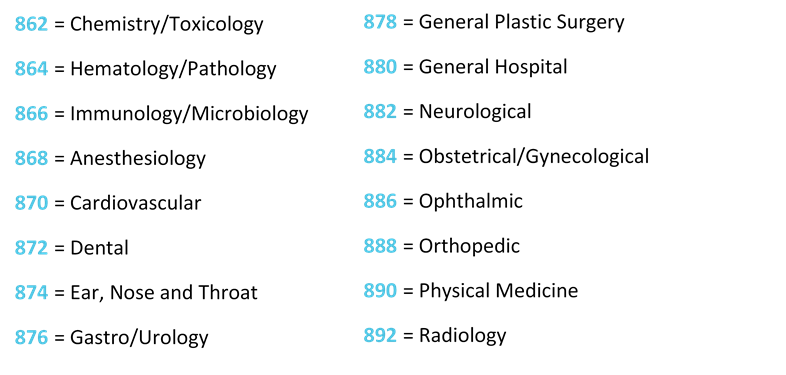
In the USA, there are 1700 generic groups of devices classified within 16 medical specialties (21 CFR 862-892). The classification is based on a risk approach. Medical devices classified as Class I are considered as having the lower risk, Class II is considered a moderate risk and class III is the higher risk.
How are the US Regulatory Pathways structured according to the classification of the device?
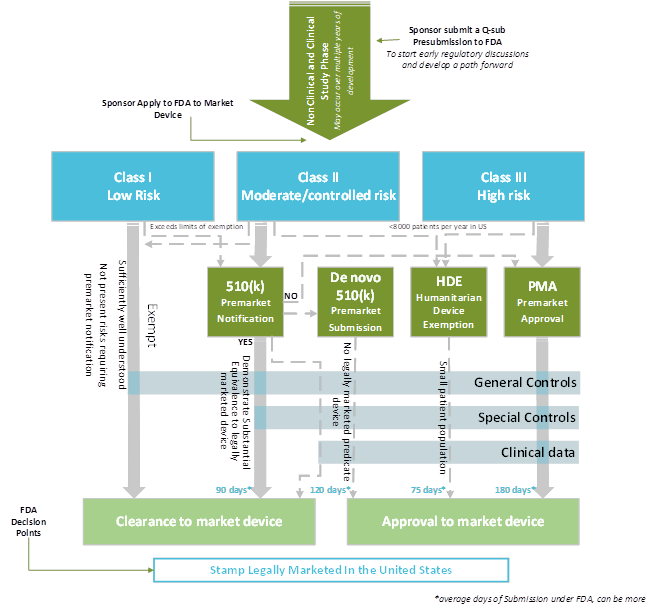
Figure 2: Structure of Medical Device US Regulatory pathway. Adapted from US legislation. Copyright 2020, Kvalito Consulting Group.
China legislation

What is the framework of China’s Legislation?

Figure 3: Framework of China’s Legislation. Adapted from Chinese legislation. Copyright 2020, Kvalito Consulting Group.
How to classify a device under Chinese regulation?
NMPA (National Medical Products Administration) takes the decision of the classification of a device via a “classification determination” procedure. Medical devices are divided into 22 categories. The classification depends on the type of device.

The NMPA (National Medical Products Administration) takes the decision of the classification of a device via a “classification determination” procedure. Medical devices are divided into 22 categories. The classification depends on the type of device.

An important point to consider for marketing a device in China:
A distinction is made between an imported device and a domestic device. On one hand, a device is considered as imported if both marketing authorization holder (MAH) and the manufacturing site are outside China and approval in the home country is required (except for device considering innovative). On the other hand, the device is considered domestic if both MAH and manufacturing sites are in China.
Imported devices need a Chinese agent who supports, among other actions, with the registration and recording of the device in China.
Author: Alix Auter, Life Science Consultant KVALITO
KVALITO is a strategic partner and global quality and compliance service and network for regulated industries. To learn more please visit us on www.kvalito.ch
If you would like to benefit from KVALITO’s expert services, feel free to send us an email to contact@kvalito.ch.
Are you looking for an exciting and challenging position as a consultant in Europe? Send us your complete application to recruiting@kvalito.ch.


