

“Discovering the unexpected is more important than confirming the known.”
—George Box, Stuart Hunter, and William G. Hunter
Introduction
The World Health Organisation (WHO) specifies that “the purpose of validation is to confirm that the computerized system specifications conform to the user’s needs and intended use by examination and provision of documented and objective evidence that the particular requirements can be fulfilled consistently.” Validation should establish confidence in the accuracy, reliability and consistency in the performance of the system and it should also ensure that all necessary technical and procedural controls are implemented confirming compliance with good documentation practices for electronic data generated by the system.
The EU EMA (European Medicines Agency) defines validation as: “the documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes” (R1). In contrast, the U.S. FDA (Food and Drug Administration) defines validation (2011) as: “the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products” (R2). The U.S. FDA has proposed a new guideline; “Computer Software Assurance for Manufacturing, Operations, and Quality System Software.” (R3). The focus here shifts away from documentation to overall tasks to be performed, to ensure that the software functions as intended and meets business requirements.
With sprawling multi-platform infrastructures, and constantly changing applications, various newly developed methodologies (service-oriented applications, business process applications, etc.), IT departments are doing their best to prepare adequately for the ever-changing world of technology. Shorter delivery times, more complex components, additional IT changes to record and control and increased risk to be managed, require an effective plan for computerized system validation and computer software assurance which enables the definition and tracking of all the validation activities needed to verify the system before being released and deployed into production.

For decades, regulated companies have been validating their computerized systems according to health authority validation principles. Does the new FDA CSA direction signify that validation will not be needed anymore, as we look ahead?
Some of you may think that the new draft FDA guideline is a paradigm shift from a pure document-focused computer system validation approach towards a more critical thinking assurance practice. However, the guideline rather explicitly clarifies that a pure documentation focus is not sufficient and not conducive for the quality of a software or product. It is indeed rational to put more emphasis on the design of the software or product, its intended use, definition, and the strategical planning as well as risk-based validation, as opposed to a taking the pure documentation creation approach. Finally, the purpose of computerized system validation is not and never has been, the creation of documentation but rather, has focused on activities emphasizing product quality, patient safety, and data integrity. Validation of computerized systems must be scientific and rational. New technology, i.e. automated testing tools or methodologies, i.e. SCRUM Agile can support the validation process. However, much more important is the predictive hazard detection and ultimately risk control of the computerized system. These validation principles are not new and have always been enforced.
Requirements Specification, Validation Planning and Reporting of GxP relevant Systems
The validation approach and the corresponding required activities can be scaled according to the nature, risk, and complexity of the business processes and the computerized system. An initial risk and impact assessment should be performed to ensure compliance with applicable GxP regulations and fitness for the intended use. Business-critical processes should be identified to determine whether the system is impacted by GxP regulations or not (including electronic records and electronic signatures).
GAMP5 is a set of guidelines for manufacturers and users of automated systems in the pharmaceutical industry, published by ISPE (International Society for Pharmaceutical Engineering). It provides a computerized system categorization to classify the system’s complexity according to 5 categories (see GAMP5 table 1 below), defining the validation and verification activities applicable to each category.
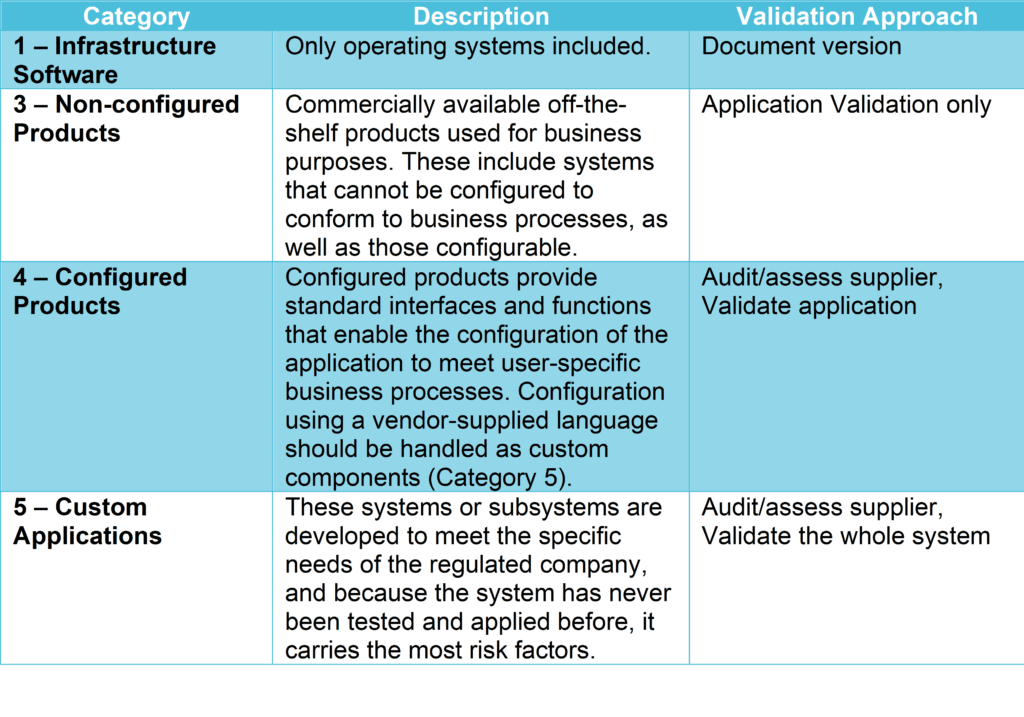
Validation effort and focus should be on aspects related to patient safety, product quality, and data integrity and therefore, business process understanding, the outcome of the vendor assessment, system complexity, the novelty of the technology, and system categorization.
For every computerized system validation, the application development process should be sufficiently planned, controlled, tested, and documented to be able to detect and correct unexpected results early enough during the software or product development. A validation plan shall be created to define validation “scope, approach, resources, schedules and the types and extent of activities, tasks, and work items” (R5). Overall, the consistency, completeness, and correctness of the software shall be demonstrated.
Since commercial off-the-shelf (COTS) software is less sophisticated than an in-house developed program, there are naturally fewer risks involved when testing the system. Although the life cycle for computerized system validation is still applied, it is less specified. The life cycle documentation produced could be less than rigorously tested in-house developed systems.
In-house applications are often designed and developed to fulfil specific business needs. Such applications are classified as GAMP Category 5. According to FDA 21CFR part 11 and EU GMP Annex 11, systems that administer, control, or support any GxP process including Manufacturing, Laboratory and Clinical Practice require validation and quality assurance. The validation and the quality assurance approach together with the related required activities can be scaled according to the nature, risk, and complexity of the system.

To ensure compliance with applicable GxP regulations and ensure fitness for intended use, initial risk and impact assessments will allow the identification of business-critical processes and the corresponding validation and quality assurance approach. If the system is impacted by GxP regulations (including electronic records and electronic signatures), as well as other regulations and requirements, such as Sarbanes-Oxley (financial), Legal, Health, Safety and Environment, Information Security and Data Privacy, validation and quality assurance is necessary to ensure regulatory compliance. Health authority regulated companies must perform computerized system validation, and they need to do it right.
Insufficient planning and a rush through the business process design might save a little money in the short term. Still, these savings will look minute and inconsequential compared to the potential costs and impacts of not spending enough time to plan and define processes. The business process must be defined before choosing and qualifying a software or product vendor.
The later you correct a mistake, the more expensive it may become (see figure 2). Therefore, each phase of the development of the software or product should be subject to detailed planning. During and between each phase of the development life cycle, a tollgate review is highly recommended before proceeding to the next stage.
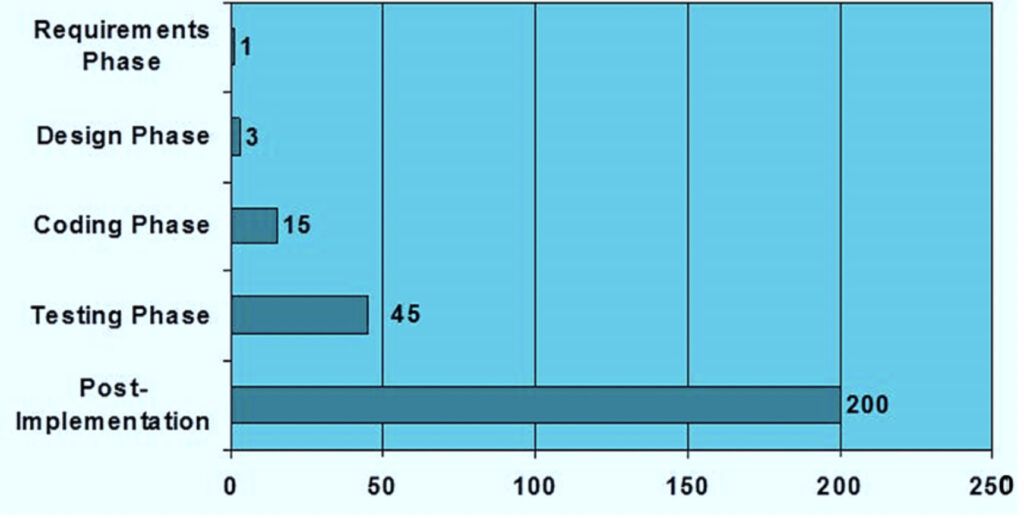
The outcome of all validation activities can be summarised in a Validation Report. Any unresolved incidents, problems, and information concerning the release of the system should be documented, and actions planned to resolve them. Impact and risk on patient safety, product quality and data integrity should be clearly included and managed.
Once the business processes are defined, the System Requirements shall be specified to ensure “the correctness and completeness of both the System Requirements and the Software Requirements” (R5). The System Requirements shall be fully accounted for through its design; hence the User Requirement Specification, Functional Requirement Specifications, and Design Requirement Specifications shall be addressed as part of the design validation phase.
Building and Development of the System
The software development process consists of dividing software work into different parts, such as design, product management, and project management. The methodology includes the definition of deliverables and artefacts.
The current process to develop software is known as Agile methodology. Additional methodologies exist, such as waterfall methodology. Agile methodology is based on iterative development, where solutions and requirements are discussed during cross-functional team meetings as continuous feedback. Within the waterfall model, the development is conducted through several phases. After the design of the computerized system is qualified, the system development can be initiated.
Traceability and Risk-based Testing of GxP relevant Systems
Testing shall be planned before execution to avoid “test quality into”. The software is coded after the test is defined, and acceptance criteria are agreed upon. Hence traceability is ensured so that user requirements are met and mapped to user acceptance test specifications.
It is not feasible to test all functions of a system with the same rigour. Testing should be performed based on documented risk evaluation. The focus of the risk evaluation shall be related to product quality, patient safety, and data integrity. However, it must be sufficiently and confidently demonstrated that the software is fit for its intended use. “To establish that confidence, software developers should use a mixture of methods and techniques to prevent software errors and to detect software errors that do occur. “The “best mix” of methods depends on many factors, including the development environment, application, size of project, language, and risk”(R5). Test efforts should be focused on the highest process and function risks. It shall be intended to identify the uncertainty in the business processes and the functionality, from which then the test strategy is developed to mitigate that risk for the system. Without knowing the risks in the business processes and functionality, the issue may be of either testing too much, or not enough, and not being able to justify the test decisions in any case. All tests must be satisfactorily executed and meet the acceptance criteria set forward. Test outcome (results) should be documented. Any unsolved critical issue with potential impact on the quality of the system and respective workarounds must be justified and a mitigation plan must be produced. Testing activities can be summarised as outlined in the sections below:
Code Reviews
A code review is a systematic examination of source code. During code reviews, the aim is to find and fix mistakes overlooked in informal test phases and improving the overall quality of the application. Code reviews ensure that the application is upgradeable as well as configurable in the future. Code reviews are performed against pre-defined coding standards and coding guidelines. “Such guidelines should include coding conventions regarding clarity, style, complexity management, and commenting.” (R5).
Installation Qualification (IQ)
The IQ verifies that all components are built, installed, and configured correctly in the appropriate operating environment and according to pre-defined IQ specifications.
Operational Qualification (OQ)/Functional testing
OQ testing is executed against functional requirement specifications to verify that all functions are properly working throughout the whole range of operativity (including upper and lower limits).
Performance Qualification (PQ)/User Acceptance Testing (UAT)
The Process Qualification (PQ) includes User Acceptance Tests (UAT), and it is recommended that PQ/UAT is executed by trained key business users to verify that the application supports business processes and user requirements specifications and therefore its intended use. It is ensuring that the system is consistently able to perform within the standard operational setup.
Go-Live Preparation of GxP- Systems
Business and IT system procedures should be available before go-live (operational processes). Business standard operating procedures ensure the correct and controlled use of the system. An operational handbook, in general, includes all related system management procedures for IT-support, including (but not limited) to release, change, configuration, and access management. A user guide should be established to ensure that the system is used correctly by its business users.
All the documents, procedures and records should be controlled, maintained, and reviewed within a defined period, ideally using a document management system. The retention periods of all computerized system validation documentation should be at least 7 years additional to the computerized system end of life cycle, even much more depending on regulation.
A training concept should be in place to ensure that only trained users with adequate training receive access to the controlled/ validated system.
After Go-Live is before Go-Live
Once the system is in use, the team responsible for keeping the software or product in a validated state needs access to all the validation documentation. It is advised to have a governance concept in place to define the way forward for escalation, decision making, and steering committee or other stakeholders involved in the maintenance of the system. Key is robust change management for continuous improvement of the system, as well as a disaster recovery plan and a business continuity plan in case the software fails to function.
In a nutshell
Computerized System Validation and Computer Software Assurance practice is beyond health authority regulation also a good practice to ensure the software or product you have created is doing what it supposed to do. Risk-based critical thinking is the basis when developing, changing, and implementing new software or products. Intended use of your software or product must be clear, and business needs must be enabled. Testing activities should be automated to avoid human errors during testing. It is also common to outsource full or part of the development to specialized vendors. Once the vendors are qualified, vendor document can be reused. Overall, your computer software assurance and computerized system validation approach should establish by objective evidence that the system meets its intended use. In addition, patient safety, product quality and data integrity must be ensured both in the development phase and in the operational phase, as you maintain your software or product.
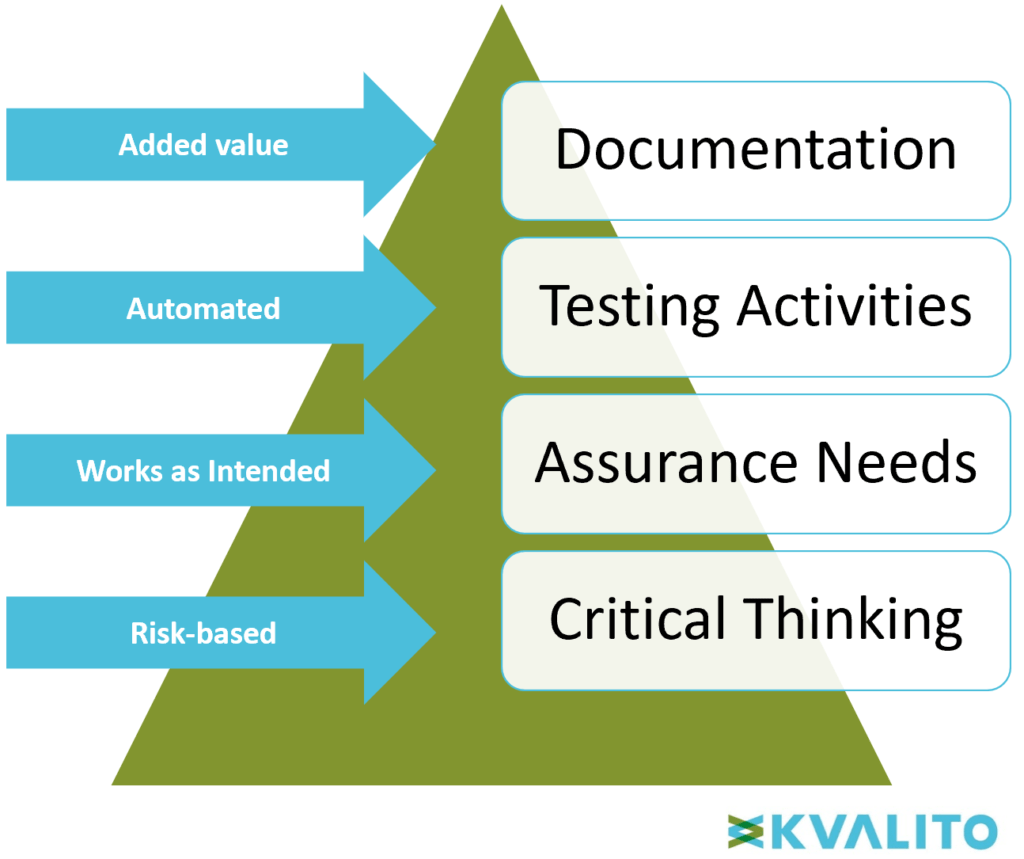
Figure 3: Computer Software Assurance Pyramid. Copyright KVALITO Consulting Group, 2020.
Read on to learn about KVALITO’s approach to mitigating risks and ensuring our clients compliance with regulations:
Validation can be some of the most confusing and challenging compliance tasks. Hiring well-trained, skilled, and experienced professionals in a timely and cost-efficient manner is prudent. KVALITO implements strategies to achieve compliance with regulatory bodies, personalizing them to meet your specific company’s needs.

As a leading niche consultancy, we interact with our clients and listen to them. This way, we make sure that our risk-based approach for validation is customized while still providing a structured and defined solution. This approach, combined with the methodology we have successfully applied on numerous international projects, will be most effective in preparing you to be ready with all the necessary documentation and proof when the health authority inspector is knocking at your door.
- We train our professionals on current health authority regulations and Good Industry Practice.
- We adhere to strict health authority rules and best practices.
- In addition, KVALITO’s focused, well-trained consultants always adhere to our clients Quality Management System.
- We assure proper identity, strength, and quality of a software or product due to support risk-based automated testing and added value documentation.
- We individually customize the testing specifications to the system’s risk categories, i.e., the more complex the system, the more rigorous the testing will be to define the capabilities of the system.
- Our consultants always keep the overview and remain in communication with all team members.
References:
R1: Guideline on process validation for finished products – Information and Data to be provided in Regulatory Submissions, European Medicines Agency, 21 November 2016
R2: Guidance for Industry Process Validation: General Principles and Practices, US FDA, January 2011
R3: US FDA Website “CDRH Proposed Guidances for Fiscal Year 2020 (FY 2020)”, https://www.fda.gov/medical-devices/guidance-documents-medical-devices-and-radiation-emitting-products/cdrh-proposed-guidances-fiscal-year-2020-fy-2020, Status: 17/Jul/2020
R4: Adler et al., GAMP5 – A Risk-Based Approach to Compliant GxP Computerized Systems, 2008
R5: US FDA, General Principles of Software Validation Final Guidance for Industry and FDA Staff, January 2002 https://www.fda.gov/media/73141/download: page 11 section 4.2, page 20 section 5.2.4
R6: Boehm Barry, Software Engineering Economics, Prentice Hall, 1981
Zowghi D. et al., The Three Cs of Requirements: Consistency, Completeness, and Correctness, April 2003
WHO, GUIDELINES ON VALIDATION – APPENDIX 5 3 VALIDATION OF COMPUTERIZED SYSTEMS 4, Working document QAS/16.667/Rev.1 ,May 2018
EMA, The European Agency for the Evaluation of Medicinal Products, Committee For Proprietary Medicinal Products (CPMP), Note For Guidance On Process Validation, CPMP/QWP/848/96, EMEA/CVMP/598/99, London 1 March 2001
WHO, DRAFT WORKING DOCUMENT FOR COMMENTS: 4 5 Guideline on Data Integrity, Working document QAS/19.819/Rev.1 June 2020
Authors: Alix Auter Life Science Consultant KVALITO and Marco Polisena, Life Science Consultant KVALITO
KVALITO is a strategic partner and global quality and compliance services and network for regulated industries. To learn more about our services, please visit us on www.kvalito.ch
If you would like to benefit from KVALITO’s expert services, feel free to send us an email: contact@kvalito.ch.
Are you looking for an exciting and challenging position as a consultant? Send your complete application to recruiting@kvalito.ch


